

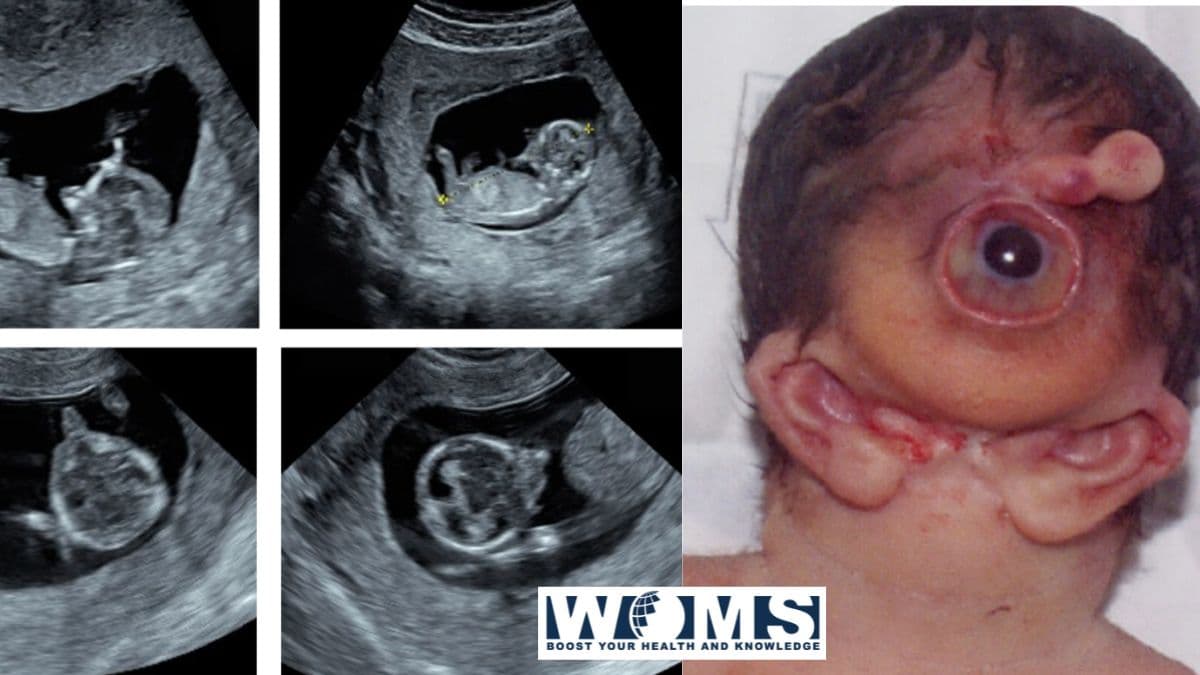
Otosefaliya bir nechta malformatsiyalar bilan bog'liq bo'lgan kam uchraydigan sindromdir. Xarakterli belgilarga mandibulaning agenezi (agnatiya) yoki gipoplastik mandibula, kichik og'iz yoki mikrostomiya, til yoki ibtidoiy tilning yo'qligi (aglossiya yoki mikroglossiya) va quloqlarning anteromedial anormal joylashishi (melotiya) kiradi. Bu malformatsiyalar, asosan, birinchi shoxli yoyning ventral qismiga tegishli. Bu odatda orqa miyadan nerv tepasi hujayralarining ko'chishi tufayli yuzaga keladi.
Bu nerv tepalik hujayralari orqa miyadan ko'chib o'tadi va maksiller va mandibulyar bo'shliqlarning rivojlanishiga sabab bo'ladi. Bu ko'zga ko'rinadigan joylar taxminan rivojlana boshlaydi Homiladorlikning 4 va 5 haftalari. Ushbu maqolada otosefaliya bilan bog'liq sabablar, taniqli xususiyatlar va patologik faktlar ko'rsatilgan. Ushbu sindrom haqida ko'proq ma'lumot olish uchun maqolani o'qishni davom eting.
Otosefaliyaning tarqalish nisbati qanday?
Otosefaliya kam uchraydigan sindrom bo'lib, faqat aholining ozchiligiga ta'sir qiladi. Ushbu sindromning tarqalish nisbati har 70 000 embriondan 1 tani tashkil qiladi. Bu, asosan, birinchi tarvaqaylab yoyining rivojlanish naqshlarining buzilishi bilan bog'liq.
Otosefaliyaning belgilari va belgilari qanday?
Yuqorida aytib o'tganimizdek, otosefaliya o'ziga xos xususiyatlarga ega, jumladan:
- Agnatiya yoki uning yo'qligi mandibula
- Malotiya yoki iyak darajasidan bir oz pastroqda joylashgan quloqlarning anteromedial holati
- Aglossiya yoki tilning yo'qligi
Bu xususiyatlardan tashqari, u tananing boshqa tuzilmalari bilan bog'liq bo'lgan turli xil belgilar va alomatlarni ham o'z ichiga oladi. Ushbu alomatlar quyidagicha:
Yuz yoki mushak-skelet tizimining o'zgarishi
- Og'izning kichik ochilishi (mikrostomiya) yoki og'izning yo'qligi (astomiya)
- Proboscis bilan siklopiya
- O'rta tanglay yorig'i bilan labda yoriq
Nevrologik belgilar
- Holoprosensefali
- Korpus kallosumning yoshi yoki malformatsiyasi
- Neyron naychalari nuqsonlari
- Anensefaliya
- To'liq aylanadigan ichki organlar (situs inversus)
- Yurak anomaliyalari
- Noaniq jinsiy a'zolar
- Ba'zi bezlarning yo'qligi
The otosefaliyani baholash tizimi bu sindromning zo'ravonlik darajasini farqlashga yordam beradi. Bu shifokorlarga bunday sharoitlarni davolashning oson usulini taqdim etadi. Sewall Rayt gvineya cho'chqalarida o'n ikki darajani tushuntirdi. Ushbu darajalar kasallikning og'irligiga qarab simptomlarni tasniflashga yordam beradi. Baholash tizimi quyidagicha:
- 1-5-sinflar embrionni agnatiya bilan (mandibulaning yo'qligi) nevrologik nuqsonlarsiz ajratib turadi.
- 6-9-sinflar og'ir holoprosensefaliyani tushuntiradi.
- 10-12-sinflarda aprosopus (yuzi va boshning katta qismi yo'qoladi) mezensefalon va prozensefalonning to'liq yo'qligi bilan namoyon bo'ladi.
Zamonaviy tarixda Rayt darajasi yuqori bo'lgan embrionning bir odamda taqdimoti haqida xabarlar mavjud.
Otosefaliyaning asosiy sababi nimada?
Otosefaliya kam uchraydigan sindrom bo'lib, asosan xromosomaning uzun qo'lidagi PRRX1 genidagi de novo mutatsiyasiga bog'liq. Avtosomal trisomiyalar odatda siklopiya kabi tibbiy muammolar bilan tez-tez uchraydi. Bundan farqli o'laroq, bu juda kam uchraydi. Bu normal homiladorlik davrining buzilishiga olib keladigan rivojlanish buzilishi.
Erta embriogenez davrida turli organlar rivojlanish jarayonlaridan o'tadi. Ushbu protseduralarning buzilishi ushbu organlarning noto'g'ri shakllanishiga olib kelishi mumkin. Birinchi novda yoyi normal homiladorlik davrining 23-26 kunida rivojlanishda davom etadi. Bu Karnegi bosqichi sifatida ham tanilgan. Oddiy rivojlanish jarayonining buzilishi faqat agnatiyaga olib kelishi mumkin. Otosefaliya bunday mutatsiyalarning kam uchraydigan natijasidir. Birinchi shox ark rivojlanib bo'lgach, bunday muammolarni davolash uchun hech qanday davo mavjud emas.
Homiladorlik haqida ko'proq bilish va homiladorlik paytida o'zingizga g'amxo'rlik qilish uchun ushbu bepul va ajoyib dasturni tekshirishingiz kerak Homiladorlik sayohati ilovasi.
Qanday boshqaruv protokollari mumkin?
Ushbu sindrom turli tuzilmalarning turli xil malformatsiyasini namoyon qiladi. Bu og'ir mikrognatiyadan siklopiya-holoprosensefali kompleksigacha bo'lgan zo'ravonlik darajasiga ega, bu odatda o'limga olib keladigan homila o'limiga olib keladi. Turli tuzilmalarning noto'g'ri shakllanishiga qarab to'rtta katta guruhga bo'linadi. Bular quyidagilar:
- Agnatiya
- Holoprosensefali bilan agnatiya
- Situs inversus va visseral anomaliyalar bilan agnatiya
- Holoprosensefaliya, situs inversus va visseral anomaliyalar bilan agnatiya
Ushbu sindrom, shuningdek, turli xil ekstrakranial anomaliyalar bilan namoyon bo'ladi, shu jumladan nerv naychalari nuqsonlari, korpus kallosum disgenezi, buyrak ektopiyasi, adrenal gipoplaziya, sefalotsel va qovurg'ayurak va vertebra malformatsiyasi.
Eng og'ir holatlar odatda o'limga olib keladi va otosefaliya bilan og'rigan homilada intrauterin o'sish sekinlashishi, erta tug'ilish yoki ventilyatsiyaning buzilishi kuzatiladi. Havo yo'llarining og'ir malformatsiyasi ham mavjud. Shu sababli, endotrakeal intubatsiya havo yo'li o'tishini tiklash qiyin. Goloprosensefali bo'lmagan atigi etti nafar bemor chaqaloqlikdan keyin tirik qoldi.
Engil agnatiya bilan og'ir bo'lmagan holatlar keng qamrovli rekonstruktiv jarrohlik muolajalar orqali boshqarilishi mumkin. Ushbu protseduralar to'g'ri yuz konturiga rioya qilish uchun mandibulani to'g'ri rekonstruksiya qilishni o'z ichiga oladi. Mushaklar etishmovchiligini bunday terapiya bilan tiklash qiyin, ammo oqilona hayot sifatiga erishiladi. Bunday bemorlar uchun yutish juda qiyin, shuning uchun enteral oziqlanish ta'minlanadi. Bunday barcha holatlar uchun oilaning to'g'ri maslahati, erta tug'ilishni rejalashtirish, traxeostomiya, dastlabki gastrotomiya va uzoq muddatli davolash rejasi semptomlarni sifatli davolash uchun zarur.
Xulosa
Otosefaliya - yuz tuzilmalarining bir nechta malformatsiyasi bilan bog'liq bo'lgan kam uchraydigan sindrom. Shuningdek, u yuzning malformatsiyasi bilan bog'liq ekstrakranial o'zgarishlarni ham o'z ichiga oladi. Bu gendagi mutatsiya tufayli yuzaga keladigan rivojlanish buzilishi. Ushbu genetik mutatsiya birinchi shoxli kamar tuzilmalarida buzilishlarga olib keladi. Bu juda halokatli sindrom bo'lib, omon qolish ehtimoli juda kam. Rekonstruktiv operatsiyalar bilan boshqarilishi mumkin bo'lgan ushbu sindromning kamroq og'ir holatlari ham mavjud.
Ba'zi prenatal anomaliyalarni tekshirish yoki diagnostikadan o'tish yaxshiroqdir
Tez-tez so'raladigan savollar (FAQ)
Tug'ilishdan oldin otosefaliyani aniqlash mumkinmi?
Prenatal tashxis juda qiyin va kam uchraydi. Hozirgacha 80 dan ortiq holatlar qayd etilgan. Aksariyat holatlar odatda otosefaliya bilan bog'liq boshqa anomaliyalar aniqlangandan keyin tasodifan topiladi. Homiladorlikning o'rta trimestrida kraniofasiyal xususiyatlarni ajratish uchun uch o'lchovli ultratovush tekshiruvi qo'llaniladi. Magnit-rezonans tomografiya, shuningdek, homilaning yuz va ko'z anomaliyalari haqida aniq ma'lumotlarni beradi.
Otosefaliya bilan chaqaloqlar omon qoladimi?
Otosefaliya - bu o'limga olib keladigan sindrom bo'lib, bu ta'sirlangan chaqaloqlarning omon qolish darajasini pasaytiradi. Omon qolgan chaqaloqlar rekonstruktiv va reabilitatsiya jarayonlarining ko'plab qiyinchiliklarini boshdan kechirishlari kerak. Bunday chaqaloqlar uchun zudlik bilan davolash havo yo'lini ta'minlash uchun traxeostomiya hisoblanadi.









![Sog’liqni saqlashda ma’naviyat kuchini ochish [PODCAST]](/_next/image/?url=https%3A%2F%2Fbackend.kasallik.uz%2Fwp-content%2Fuploads%2F2023%2F05%2FUnveiling-the-power-of-spirituality-in-health-care.jpg&w=3840&q=75)
Javoblar (0 )